Current Topics
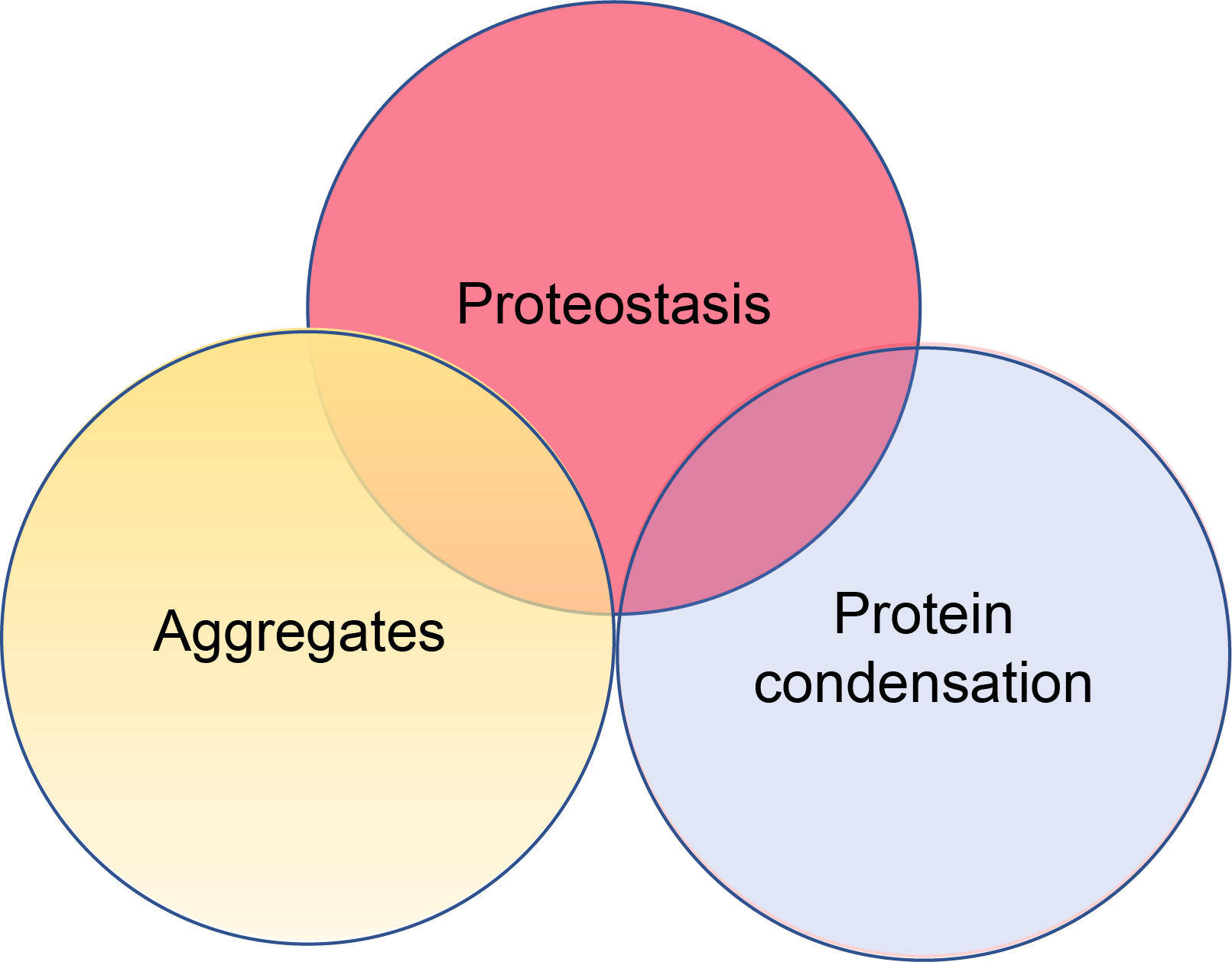
Our lab investigates the role of ubiquitin(Ub)-proteasome system (UPS) in proteostasis and cell stress. We are especially interested in the clearance mechanisms for pathological aggregates in models cell lines and neurons and how malfunctions of the proteasome system leads to proteotoxic stress, inflammation and eventually degeneration of physiological functions. Our central question is: how do the UPS and proteostasis systems target pathological aggregates, regulate cell stress and proteotoxicity? We address this question using a multi-disciplinary approach combining 1) advanced imaging in live neuronal models and 2) biochemical studies of aggregate disassembly mechanisms and stress responses. We are now seeking novel ways to restrict the build-up of protein aggregates and the progression of neurodegenerative disorders. We are especially interested in examining proteostasis in tauopathy and alpha-synucleinopathy models. To achieve this, we use CRISPR genetic editing of IPSC-differentiated cell models with pathological tau and alpha-synuclein backgrounds, perform biochemical examinations of tau, alpha-synuclein proteins and characterise the molecular features of their aggregates in situ. More recently, our research has taken us in a novel direction to investigate the mechanisms of protein condensation that may be proteostasis processes when targeting protein aggregates in cells. Our observations are crucial and at a critical stage because protein condensates have gained increasing attention as an important mechanism in cell biology, but the distinct physical principles and biological consequences of phase separation are not well-studied. Using pathologically originated aggregates to study proteasomes in neurons, our research will establish model systems to study their mechanisms and proteostasis responses in healthy- versus disease state.
Plain English
Our lab works on uncovering the events that occur within and between cells found in the human brain and how these cellular communications may behave abnormally in dementia and neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease (AD and PD, respectively). A major component of all our cells are proteins, which are responsible for catalysing reactions, provide scaffolding for e.g. changes in cellular structures and to maintain the cell in a healthy state.
Proteins that become obsolete or dysfunctional are removed by molecular machines called “proteasomes”, which recycle proteins back into their chemical components. Some obsolete proteins are ‘sticky’ in nature and if left unchecked, they tend to stick together and accumulate to form toxic agents that harm cells, a process known as “protein aggregation”. Proteasomes remove these obsolete proteins and are therefore important to prevent the build-up of toxic agents. The consequences of these toxic agents can be dire, causing stress within nerve cells so that normal biological communication cannot occur and eventually result in their death, known as “neurodegeneration”. Therefore, understanding the function of proteasomes is important in regulating biological processes through changing the level of proteins involved in these processes.
Current Research Interests
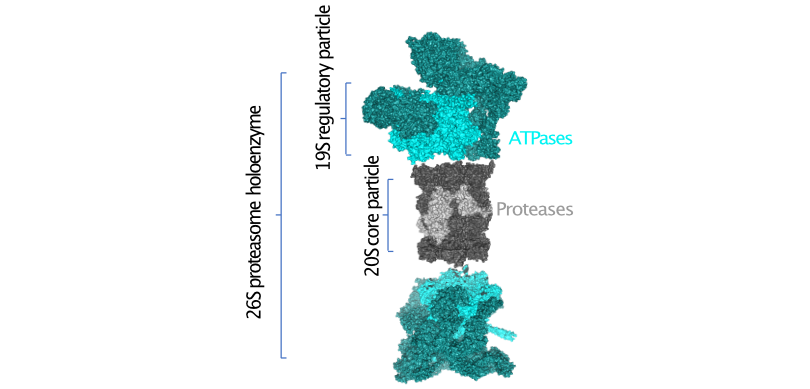
Inducible proteasome systems in AD
Proteasomes are abundant molecular recycling machines that govern a wide range of important processes such as proteostasis, pro-inflammatory signalling, DNA repair and various cell stress responses. The canonical proteasome holoenzyme consists of a proteolytic 20S core particle, capped on one or both ends with 19S regulatory particles, which recognise and unfold substrates. Besides these constitutively expressed particles, other specific particles exist, such as the nucleus-specific PA200, as well as the immune-activated 'immunoproteasome' and the 11S REG. Emerging data have suggested that subunits of the immunoproteasome are AD risk genes and that they are found near protein aggregates in AD models. Our lab is interested in the various proteasome particles that exist in different cell types and how their abundance is influenced by neurodegeneration. We are studying the activity and localisation of proteasome particles involved in cell stress repsonses.

Examination of pathological aggregates
We previously reported that alpha-synuclein aggregates from PD or DLB that are smaller than 450 nm in size penetrate their plasma membrane and are subsequently engaged by a dense entity of concentrated proteasomes. We also observed that the concentration of aggregates defined by this size range correlated linearly with toxicity level determined by lactate dehydrogenase (LDH) assay. Our suggested that the toxicity profile generated appeared to be disease-specific; that is, within the aggregate size range, PD aggregates was found to be more toxic than DLB aggregates. Based on this, we hypothesise that a fraction of disease-specific aggregates is present in each dementia type and that this fraction may be used for disease identification. We already have developed deep learning-based computational approaches for classification (patent application GB2214417.4) based on analysis of aggregates from postmortem brain tissues. We are now expanding these approaches to four different dementia types to determine their suitability for early-stage disease detection.
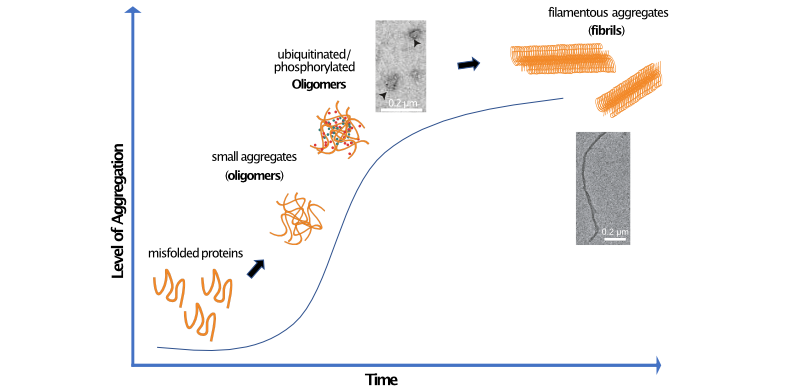
Reversing protein aggregation
The presence of insoluble protein deposits in the brain is a hallmark of neurodegenerative disorders, which include Alzheimer's and Parkinson's disease. Distinct misfolded proteins, such as tau and alpha-synuclein, have been associated with different neurodegenerative disorders. Aggregation of these misfolded proteins goes through several stages, during which the monomeric proteins assemble into oligomers in solution, which then through further aggregation and conformationally re-arrangements eventually form filament structures (fibrils). While the aggregation process has gained much attention, relatively little is known about the reverse process of aggregate removal. Our lab is interested in which additional co-proteins may assist proteasomes in removing stable fibrils and how the toxic oligomers that damage biological functions may be removed by the ubiquitin-proteasome system (UPS), thus reversing neurodegeneration. We are now expanding our research into understanding how additional modifications may influence aggregation, toxicity and susceptibility to be targeted by the UPS.
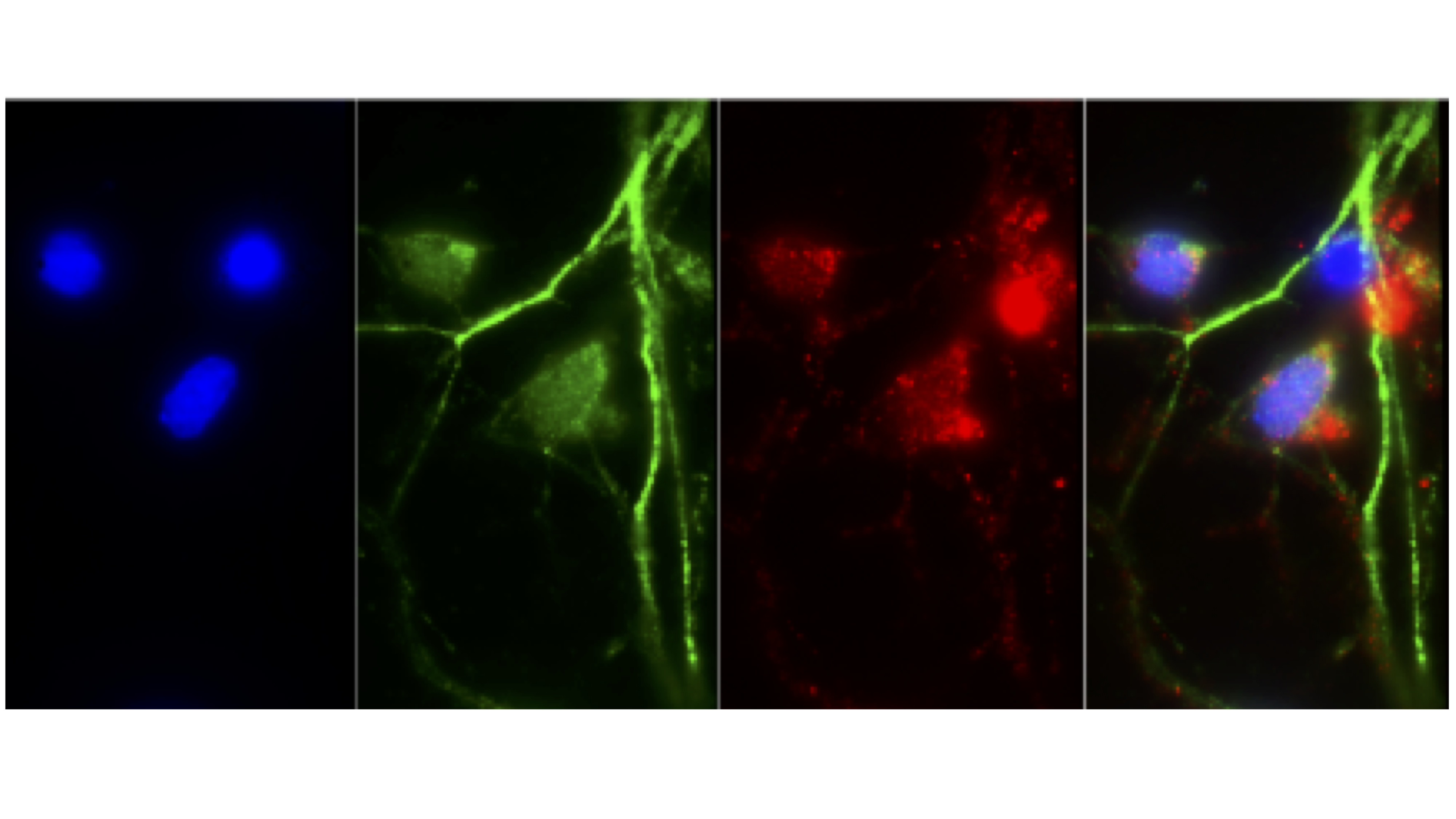
Protein phase separation and condensation
Increasing evidence suggest that proteasomes may concentrate into membraneless LLPS-dependent foci bodies in response to stimuli such as starvation or osmotic stress. This suggests that proteasomes may redistribute during stress, likley altering proteasome-regulated processes and general proteostasis. We recently postulated that cellular damage by aggregates of misfolded proteins may also induce such proteasome condensate, which we named transient aggregate-associated droplets (TAADs). These TAADs would facilitate proteasome interaction with their co-proteins (e.g. chaperones, proteasomal shuttle factors, ubiquitination enzymes) to accelerate aggregate removal, thus reducing proteotoxicity. Our PNAS paper (Morten et al. 2022) further provided evidence for TAADs, which colocalised with invading alpha-synuclein aggregates within 12 hrs, eventually leading to reduced aggregate abundance. We now seek to identify mechanisms that drive TAAD formation.
Imaging Techniques

Advanced Microscopy
Our lab has built a fully automated azimuthal- or spinning total-internal reflection fluorescence (TIRF) microscope with 2D and 3D super-resolution imaging capabilities. This provides sub-millisecond detection of rapid processes in live cells and protein localisation in fixed cells.

Data Analysis
We have a portfolio of analysis methods which include custom-written codes for single-particle identification and trafficking, protein co-localisation, cell masking and sub pixel aggregate size analysis.
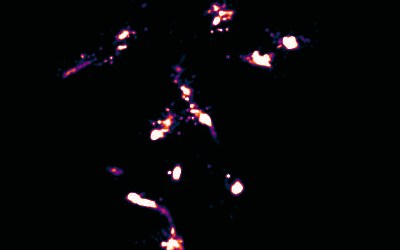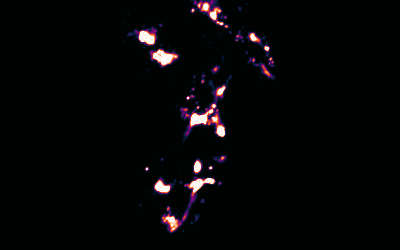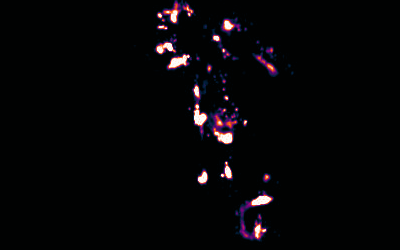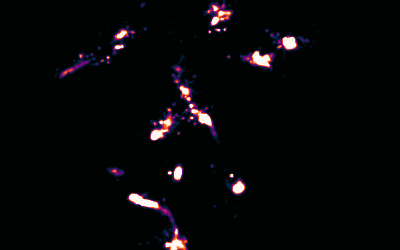
Image Reconstruction
We have recently built on previous methods to reconstruct multicolour super-resolution images in 3D from within cells and tissues.
Experimental Systems

Molecular Biology
The lab uses a range of canonical molecular biology and protein biochemistry techniques for in vitro studies.

iPSC Cultures
Our lab uses tauopathy and synucleinopathy patient derived iPSC lines. We are further differentiating iPSC-neurons and glia using established protocols.
CRISPR/Gene Modifications
We are modifying the genome to enable live imaging of specific proteins in neurons and microglial cultures. Using CRISPR knockouts we are investigating the function of specific proteasome-related genes.

Mouse and Human Tissues
In collaboration with other groups within the UK DRI, we are studying different neurodegenerative diseases from aggregate-containing models and patient donors.